(1 国家食品药品监督管理总局药品审评中心,北京100038;2国家食品药品监督管理总局食品药品审核查验中心,北京100061)
[摘要] 本文主要以相关指导原则和电子刊物为基础,结合QbD理念及多肽药物特点,探讨多肽制备工艺中起始物料、工艺研究优化、中间体控制等环节对产品有关物质的影响,实现对多肽药物有关物质的有效控制。
[关键词] 合成多肽;制备工艺;过程控制;有关物质
[中图分类号] R95 [文献标志码] C [文章编号] 1003-3734(2017)18-2143-06
Effect of manufacturing process and process control on related substances of synthetic peptide drugs
Hu Yuxi1 Jiang Yu1 Han Tianjiao2
(1 Center for Drug evaluation, China Food and Drug Administration, Beijing 100038, China;2 Center for Food and Drug Inspection, China Food and Drug Administration, Beijing 100061, China)
[abstract] Referencing to relevant guidelines and electronic journals, this paper mainly discusses the effects of starting materials, process optimization and intermediates control on the quality of synthetic peptide drugs, especially related substances, based on QbD concept. The objective is to reduce related substances of synthetic peptide drugs through enhancing the process control.
[Key words] synthetic peptide; manufacturing process; process control; related substances
[概述] 多肽药物具有生物活性高、特异性强、毒性反应弱、在体内不易产生聚集、与其他药物相互作用比较少、与体内受体亲和性高等优点,是国内外制药企业研发热点之一,尤其随着微球、脂质体、聚乙二醇(PEG)修饰等方法的开发,解决了多肽药物稳定性差,体内易降解,半衰期短等成药性差的问题,在促进多肽新药开发的同时,也促进了多肽药物的二次开发利用[1-2]。
国家食品药品监督管理总局于2007年10月发布了《合成多肽药物药学研究技术指导原则》[3],黄晓龙等[4-7]和张哲峰等[8-9]对指导原则和相关共性问题进行解读和探讨。美国药典会在2013年成立了多肽专家小组,依据目前可用的或将来可能采取的监管规章制度,对合成多肽的质量属性进行评估,目前已经发表了多篇专论,涵盖了多肽药物合成工艺、质量研究等内容[10-12]。上述文章和专著的发表,对多肽类药物的开发起到了积极的指导和参考作用。
多肽类药物合成过程中可能生成差向肽、缺失肽和错结肽等杂质,很多杂质与目标产物结构、化学性质非常相近,仅采用经典小分子化学药物制备过程中常用的萃取、重结晶等提纯手段无法得到合格产品,需经过复杂的提纯过程才能得到合格产品。有关物质对多肽药物的安全性、有效性具有直接影响,是产品的关键质量属性,同时也是产品开发和技术审评关注的重点内容。在技术审评过程中,发现有关物质研究深度差异较大。如某10肽药物,研究了69个杂质,其中差向肽杂质29个,缺失肽杂质18个,错结肽杂质5个,降解杂质17个;某28肽药物,仅研究了端位差向肽杂质。如果多肽药物含有N个手性氨基酸,理论上差向肽杂质为2N个,再结合缺失肽、错结肽和降解杂质等,需考察的潜在杂质数量巨大,逐个研究不具有可操作性。因此建立从源头控制产品质量的手段,加强过程控制,可有效降低终产品有关物质分析和控制难度,降低产品质量风险。
1、多肽类药物概况
近30年来,学术界对多肽类药物的关注度持续升高,涉及多肽类药物研究各个方面的论文发表数量也持续上升。Web of Science数据库中,多肽类论文发表数量从8081篇(1980-1984年度)升至103426篇(2010-2014年度)。学术界持续升高的关注度也已转化为持续扩张的多肽类药物市场,目前全球已经批准了近100个多肽产品上市,2015年全球销售总额近220亿美金,占医药品市场总份额的2%左右。多肽药物市场涌现出了一系列年销售突破十亿美金的重磅产品,如Glatiramer(格拉替雷),Liraglutide(利拉鲁肽)、Leuprorelin(亮丙瑞林)等。
多肽类药物在治疗肿瘤、糖尿病、心血管疾病、肢端体肥大症、骨质疏松症、胃肠道疾病、中枢神经系统疾病、免疫疾病以及抗病毒、抗菌等方面具有显著的疗效,目前多肽类药物研发项目大多集中在抗癌、糖尿病、抗心力衰竭等领域,据不完全统计,活跃在临床III期及以后的多肽药物有以下十几种:Semeglutide、Ularitide、Plitidepsin、Abaloparatide、Selepressin、Difelikefalin、Bremelanotide、Afamaelanotide、Paclitaxtide、Thymosin Beta-4、Tyroserleutide、Sifuvitide、Batifiban、Zoptarelin Doxorubicin、Aviptadill、Paclitaxel Poligumex、Reltecimod、Disomotide等。随着一些潜在重磅药物的上市,多肽药物市场依旧可以保持良好的增长势头。
2、合成策略选择
多肽的制备方法有生物组织提取法、基因重组表达法和化学合成法,目前已上市的多肽药物有90%以上可通过化学合成法制备。多肽药物主要由氨基酸或者氨基酸衍生物组成,可以通过氨基酸逐步连接或者多个肽链片段缩合,再经过较复杂的纯化工艺得到。目前化学合成多肽主要包括液相法、固相法、液相固相结合法。
1907年Fischer成功地采用液相法合成十八肽Leu- (Gly)3- Leu- (Gly)3- Leu- (Gly)9,至今已有近百年的历史。液相多肽合成可以节约原料、定量监控反应进度,但是其中间体纯化操作复杂,后处理繁琐造成合成周期较长、产品损失较大。1963年,Merrifield创立了将多肽C端的氨基酸固定在不溶性树脂上,然后在树脂上依次偶联氨基酸合成多肽的固相合成法(Solid-Phase Peptide Synthesis,简称SPPS),固相合成操作简便,通过快速过滤、洗涤未反应的中间体/原料,避免液相合成多肽中间体纯化的步骤,从而减少中间纯化损失,提高产品总收率。同时,固相合成法具有便于计算机控制、可实现自动化操作等优点,在药物开发、蛋白质结构研究、免疫学研究等领域表现出了显著的优越性,从此多肽合成进入了一个飞速发展的阶段。
固相合成法是将氨基酸C端固定在树脂上,再经过与目标氨基酸反复偶联,肽链的偶联过程涉及到一连串的“缩合”-“脱保护”等重复操作,在缩合步骤中,通常使用过量的活化氨基酸以确保N端氨基酸完全反应,这些氨基酸通常使用“临时性”保护基保护N-α氨基,如果氨基酸的侧链也有反应活性,则需要使用“半永久性”保护基保护起来。缩合完成之后,N-α氨基进行选择性脱保护同时保留肽链的侧链“半永久性”保护基,释放出N-α氨基以备后续连接。合成目标序列多肽后,利用催化剂脱除“半永久性”保护基,同时将肽链与树脂裂解,再经过纯化等操作步骤制备目标多肽。
按照氨基酸保护策略,固相多肽合成法可分为Boc(叔丁氧羰基)合成法和Fmoc(9-芴甲氧羰基)合成法。Boc合成法采用三氟乙酸(TFA)可脱除的Boc基团为α-氨基保护基,侧链保护采用苄醇类,最终脱保护需要使用腐蚀性较强的氢氟酸(HF)或者对三氟甲基水杨酸(TFMSA)。Fmoc法采用碱可脱除的Fmoc为α-氨基保护基,侧链保护采用TFA可脱除的叔丁氧羰基等,由于Fmoc法避免了中间体反复酸处理过程,同时避免了腐蚀性较强的强酸处理过程,可有效降低副反应的发生,为目前固相法合成多肽的首选方法。
除以上液相法和固相法外,还有固相液相结合法,主要针对特定长肽序列。序列超过50个氨基酸的长肽合成,若采用氨基酸逐步偶联的固相法制备,随着氨基酸数量的增多,空间位阻等因素会使反应难度加大,具有一定的局限性。可将多肽序列分成多个短肽片段,先用固相法合成短肽片段,再采用液相法将短肽片段逐一接连起来。这种方法可以通过提高短肽片段的纯度,来降低最终产物的杂质含量,尤其与终产品结构、性质相似的杂质可以得到有效控制,从而降低提纯难度,达到提高产品质量和收率的目的。
多肽药物往往因为其独特的多肽序列,可能存在数条合成路线,每条路线均有其各自的特点。开发多肽药物前期应充分调研,掌握药物特性,通过工艺摸索选择最适合特定产品的工艺路线。
例如某9肽药物,在早期开发过程中采用固相合成Fmoc保护策略,由C端到N端逐步偶联目标氨基酸的方法。在研究过程中发现4、5位氨基酸链接困难,会产生一定量的缺失4位、5位、4和5位氨基酸的缺失肽杂质,其中单氨基酸缺失肽杂质在后续提纯过程中很难除去。通过改变合成策略,利用固相合成1-4和5-9肽链片段后,再进一步偶联成目标产物,可有效减少缺失1-2个氨基酸的缺失肽杂质的生成,反应体系中所残留的主要杂质为与目标产物性质相差较大的1-4和5-9肽链片段,可有效降低提纯难度。
目前,多肽药物的制备主流方法为固相合成Fmoc保护法,本文主要针对该制备路线相关工艺过程控制进行论述。
3、起始物料控制
International Conference on Harmonization(ICH)Q11中强调了对起始物料的监控,特别是起始物料选择的合理性,还着重说明了起始物料杂质谱研究的必要性,了解它们在生产过程当中的作用,以及它们与最终API中杂质之间的关系。
多肽合成中,各种保护氨基酸作为构成终产品的结构片段,其质量情况直接影响终产品的质量,应定义为关键起始物料,并建立严格的质控标准。对于保护氨基酸企业制定的内控标准中,除常规的性状、比旋度、纯度项目以外,还应该开展杂质谱研究。保护氨基酸杂质谱研究应包括游离氨基酸、对映异构体、二聚体或寡肽、Fmoc氨基酸中含有β-Ala类杂质、其他非目标氨基酸等。
构成多肽药物的氨基酸绝大多数含有手性中心,对映异构体杂质引入肽链后,会形成结构、性质与目标产物相似的差向肽杂质,提纯难度较大。需利用手性HPLC或其他灵敏度高的分析方法控制起始物料中潜在的对映异构体杂质,并建立严格的质控要求。
对保护氨基酸其他杂质的分析,首先应结合相关文献或其他研究资料,根据氨基酸自身性质进行杂质分析,如Fmoc-Leu-OH中可能含有Fmoc-IlE-OH杂质,Fmoc-Ala-OH和Fmoc-Pro-OH中可能含有相应的二聚体杂质。
其次,需结合制备工艺过程进行研究,如在采用N-9-芴甲氧基羰基氧琥珀酰亚胺(Fmoc-OSu)制备Fmoc-保护氨基酸时,会伴随Fmoc-β-Ala和Fmoc-β-Ala-AA生成,如图1。反应混合物中的亲核试剂进攻Fmoc-OSu中的一个羰基后,通过Lossen重排即得到β-Ala结构,形成β-Ala氨基酸残基,后者可通过另一当量Fmoc-OSu发生Fmoc-保护,并生成Fmoc-β-Ala杂质。拟发生Fmoc-保护的游离氨基也可与生成的β-Ala氨基酸残基反应生成二肽,后者可发生Fmoc-保护从而生成Fmoc-β-Ala-AA。

图1 多肽类药物起始物料Fmoc-β-Ala-OH和Fmoc-β-Ala-AA-OH杂质的生成机理。
在充分掌握了保护氨基酸潜在杂质谱后,需开发出能有效检出各潜在杂质的分析方法,并按照相关技术指导原则要求,对分析方法进行全面的方法学验证,充分证明拟采用方法的可行性。如某申报资料Fmoc保护氨基酸检测报告中,HPLC法测定纯度为为98.9%,滴定法测定含量仅为92.1%,产品中可能包含未被检出的杂质,有关物质分析方法可能存在缺陷。
保护氨基酸中可能包含大量可引入到终产品中的杂质,如对映异构体杂质,生成的差向肽杂质性质与目标产物接近,提纯精制过程除去困难;某些由保护氨基酸引入的杂质,反应活性远高于目标保护氨基酸,会大量引入到终产品中,造成多肽药物杂质谱复杂,分析困难。因此加强保护氨基酸的杂质谱分析,并建立严格的质控标准,对降低终产品提纯难度、提高终产品质量具有积极意义。
4、工艺研究和优化
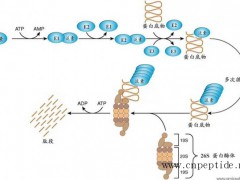
多肽药物工艺研究和优化过程与小分子药物差异较大,多肽类药物如果对每个氨基酸偶联反应步骤所涉及的反应溶剂、催化剂、反应时间、反应温度等条件进行逐一摸索,工作量巨大且没有可操作性。因此,需结合多肽药物组成的每个氨基酸结构和性质特点,进行有针对性的考察,如组氨酸(His)、半胱氨酸(Cys)、苯丙氨酸(Phe)等易消旋,天冬酰胺(Asn)、谷氨酰胺(Gln)等易水解,蛋氨酸(Met)、半胱氨酸(Cys)、组氨酸(His)、色氨酸(Trp)等易氧化,天冬氨酸(Asp)参与形成的肽链较易断裂,尤其是天冬氨酸-脯氨酸(Asp-Pro)和天冬氨酸-甘氨酸(Asp-Gly)肽键。分析并考察多肽药物在合成过程中保护氨基酸的活化和偶联、脱保护、肽链裂解、修饰(二硫键链接)、多级纯化、酸根转型、浓缩、冷冻干燥等各操作单元中可能产生的杂质,建立相应控制策略,减少杂质的产生。图2为在多肽制备过程中可能产生的杂质情况。[12]
图2 多肽工艺流程及可能产生的杂质
工艺研究和优化过程,能反应出对工艺的理解和掌控程度,在工艺考察过程中应对如下内容进行考察,最大限度的降低杂质的生成:
4.1 氨基酸偶联过程
固相法合成多肽,在氨基酸反复链接的过程中,因肽链未与树脂裂解,缺少有效的中控手段,因此在工艺开发过程中参数的摸索显得尤为重要。可对合成中间态进行研究,设定目标中间肽数个(如每5个肽为一个循环),在工艺研究中对中间肽进行检查,如增重、质谱、氨基酸序列和纯度等,充分研究每个氨基酸连接难易程度及在反应过程中可能发生的消旋等副反应,建立针对性的控制手段。如某28肽药物,在中间肽研究过程中发现12位和13位氨基酸偶联反应进行不完全,通过优化催化体系、氨基酸投料比等均不能使反应进行完全,因此合成工艺中在上述氨基酸偶联结束后采用了乙酰化封端处理,使非目标肽链不参与后续偶联反应,最终与树脂裂解之后生成的被乙酰化的多肽杂质由于与目标产物氨基酸个数相差较大,通过拟定的提纯手段可以有效除去。如不进行封端处理,未反应完全的活性链段可能参与后续反应,进而生成系列缺失肽杂质,部分杂质性质与目标产物性质相近,影响终产品提纯效果。通过对反应中间肽的研究,尤其是较长的肽链,可揭示反应过程中不容易控制的质控点,通过对反应条件和合成策略的优化,可以降低终产品中杂质的含量,降低提纯难度。该重复链接过程还应关注以下方面:
与树脂链接:氨基酸在与树脂链接反应前,应使树脂充分溶胀,反应结束后应考察树脂反应活性位点是否完全反应,未反应的活性位点应采取封端处理,否则会在产品中引入C端缺失肽杂质。
偶联反应溶剂:一般多肽合成中,常用的反应溶剂为二甲基甲酰胺(DMF)、二氯甲烷(DCM)等,不同溶剂的溶解能力不同,DMF溶解度好,是多数固相合成的常用溶剂,但DMF介电常数高,容易诱导引发消旋反应发生,DCM属于低介电常数的溶剂,能降低该副反应发生,但存在溶解性差的问题。因此在氨基酸偶联溶剂中,需综合判断采用单一溶剂还是混合溶剂进行偶联反应。
缩合剂的选择:目前所开发的氨基酸偶联缩合剂种类繁多,不同缩合剂的缩合效率、价格差异较大,部分氨基酸在偶联的过程中容易发生消旋反应,采用高效缩合剂可有效避免消旋反应的发生,应结合氨基酸反应活性和特点,说明所选用缩合剂的合理性。关于缩合剂种类和催化效果已有较多文章进行说明,在此不进一步赘述。需要说明的是偶联反应所用缩合剂多为碱性试剂,反应过程中的肽链在高浓度碱性体系中可能发生消旋等副反应,因此合理的缩合剂使用浓度不仅可有效降低成本并可提高产品质量。
反应终点判断:缺失肽为整个多肽序列中缺少一个或多个氨基酸残基的杂质,产生于合成过程中活化的氨基酸连接不完全或者N端氨基脱保护不完全。目前,固相合成中多用“Kaiser 比色测试”(茚三酮检测)来检测反应是否完全,但该方法不能定量检测,因此也不能完全避免缺失肽杂质的生成。当缺失的氨基酸是甘氨酸(Gly)或丙氨酸(Ala)等结构简单氨基酸时,生成的缺失肽杂质提纯清除困难。在这种情况下,利用前文所述的中间肽考察策略则非常必要,对易产生缺失肽步骤建立更严格的工艺规程。同时考虑采用Kaiser试验、三硝基苯磺酸(TNBS)试验或其他不同的检测方法,检测结果相互佐证,避免假阳性/假阴性结果,以保证反应终点判断的正确性。
树脂的洗涤:固相法合成多肽过程中,为确保偶联效率最大化,每步偶联过程需加入过量的Fmoc-保护氨基酸,但如有部分残留并引入后续偶联步骤,则可能导致错结肽杂质的形成。因此,保证每步反应树脂洗涤的充分性,具有重要意义。需建立相应的分析方法,并对拟定方法的灵敏度等进行方法学验证,研究氨基酸偶联过程树脂洗涤的充分性。
目前申报资料中对氨基酸偶联过程的摸索较薄弱,如某28肽药物,从1~28位氨基酸偶联反应溶剂、催化剂、温度等条件均相同,仅按氨基酸序列改变每步反应使用的保护氨基酸。在氨基酸偶联过程中,由于氨基酸活性、空间位阻等因素的影响,氨基酸偶联难度均不同,反应条件也应该有所差异,需进行相应的针对性研究,证明工艺的合理性。
4.2 裂解条件的选择
完成全保护肽序列的合成之后,通过酸解作用将侧链“半永久性”保护基除去,同时将肽链从树脂上裂解下来。在酸解过程中,“半永久性”保护基和树脂链接臂会生成三苯基甲基、叔丁基等碳正离子,在此过程中需要使用一种或多种清除剂来捕获产生的碳正离子,以避免其与敏感氨基酸侧链发生反应而产生杂质(如酪氨酸、丝氨酸和苏氨酸上的羟基)。上述碳正离子清除剂通常包括巯基化合物(如1,2-乙二硫醇,2-巯基乙醇等)、酚类化合物(如苯甲醚、对甲苯酚等)和水。此外三异丙基硅烷等硅烷衍生物也显示出良好的叔丁基、三苯基甲基等碳正离子俘获能力。需根据在反应过程中可能生成的碳正离子类型,选择合适的清除剂配方组成。
某多肽药物裂解液配方摸索过程如表1,可以看出利用不同裂解液配方制备的产品纯度差异较大,尤其是与主峰接近的杂质含量差异较大。通过优化裂解液配方组成不仅可以提高产品纯度,也可以降低后续提纯步骤难度。
表1 不同裂解液配方组成对产品质量的影响
4.3 肽链的修饰
部分多肽药物合成目标氨基酸序列后,需进一步结构修饰,以得到目标产物。以二硫键的构建为例,形成二硫键的方法有很多种,如空气氧化法、碘氧化法等,每种方法均有各自特点,应对所选择的氧化方法的合理性进行充分说明。二硫键链接过程中可能产生未完全氧化杂质,过度氧化成亚砜、砜等杂质,二硫键链接位点错误等杂质,应对反应参数进行详细研究,确定工艺条件的合理性。
4.4 纯化
多肽药物合成一般需采用多种不同原理的HPLC提纯方法进行精制,如反向、离子交换、分子排阻HPLC方法等,在相关指导原则和电子刊物[3,4,7]中已经对采用不同提纯方法的必要性进行了充分说明,在此不做进一步赘述。
在提纯过程中应考察提纯工艺去除缺失肽、错结肽、差向肽等杂质的效果,并对精制前后样品的进行质量对比。在提纯工艺描述中应明确色谱柱、填料规格,上样量,收集产品纯度范围,超出纯度范围产品是否进行二次纯化,二次纯化条件等。应根据GMP要求,建立规范的纯化工艺流程。
4.5 富集
多肽药物一般采用冻干手段制备最终产品,部分多肽药物对热不稳定,在开发冻干工艺时需充分考虑冻干条件的合理性,并对冻干前后样品进行质量对比,验证拟定工艺不会对产品质量造成明显影响。由于冻干过程中可能对多肽药物产生不良影响,目前也有部分多肽原料药采用沉淀法或喷雾干燥法富集,如采用后两种富集手段,应充分考察方法的适用性和合理性。
如某多肽类药物对热不稳定,初次申报时拟定二次干燥温度为40℃,时间8小时,审评时对该部分内容提出异议,要求申请人提供冻干前后质量对比资料,必要时对冻干条件进行优化。经申请人研究,发现产品在超过30℃条件下放置,有关物质增加明显,最终将二次干燥工艺修订为25℃,时间6小时。
5、中间体控制:
固相合成多肽药物,在肽链与树脂裂解前,缺少有效的中间体控制手段。因此当多肽与树脂裂解后,应建立严格的中间体控制措施。需结合工艺开发过程中易产生的杂质情况,关注潜在的缺失肽、错结肽、差向肽等杂质的残留情况,综合分析后续提纯、转盐、冻干等步骤的杂质谱变化情况,建立相应的质控手段。
[小结] 多肽药物杂质谱的复杂性,决定了该类药物不能仅靠终产品提纯、样品检测控制产品质量,需加强工艺过程控制。上述例子不能涵盖多肽药物合成工艺研究过程中每一个细节,工艺研究应结合多肽药物组成氨基酸的特点,进行针对性的研究,建立相应的过程控制策略,最大限度的降低潜在的缺失肽、错结肽、差向肽等杂质的生成,达到有效控制产品质量的目的。
参考文献:
[1] 王姗.热敏凝胶用于多肽与蛋白药物的研究进展[J].中国新药杂志 ,2014,23(19):2266-2270.
[2] 程念.多肽药物长效化研究进展[J].中国新药杂志 ,2016,25(22):2574-2580.
[3] 国家食品药品监督管理总局.合成多肽药物药学研究技术指导原则 [S].2007.
[4] 黄晓龙.美国FDA关于合成多肽的指导原则[J].中国新药杂志2001,10(8):626-628.
[5] 康建磊,徐冰珠,李建宇.关于合成多肽药物中非对映异构体杂质的研究[J].中国现代应用药学,2010,27(5):387-389.
[6] 王鹏.合成多肽结构确证和质量研究[J].中国新药杂志 ,2009 ,18(24):2302-2305.
[7] 王鹏.合成多肽药物的合成工艺中关键问题分析[J].中国新药杂志 ,2010 ,19(2):102-105.
[8] 张哲峰.合成肽类药物质量研究与质量标准工作的几点考虑 [EB/0L].(2006-11-10).http://www.cde.org.cn/dzkw.do?method=largePage&id=1520.
[9] 国家食品药品监督管理总局药品审评中心五部 .合成多肽药物有关物质研究的几点考虑[EB/0L].(2007-11-27)http://www.cde.org.cn/dzkw.do?method=largePage&id=2353.
[10] EGGRN I.Control strategies for synthetic therapeutic peptide APIs Part I:analytical considerations[J].Biopharm Intern, 2014,27(3):16-21.
[11] EGGRN I.Control strategies for synthetic therapeutic peptide APIs Part II:Raw material considerations[J].Biopharm Intern, 2014,27(4):24-27.
[12] EGGRN I.Control strategies for synthetic therapeutic peptide APIs Part III:manufacturing process considerations[J].Biopharm Intern, 2014,27(6):42-46.